低黏度柔性UV 固化环氧丙烯酸酯的合成及性能研究
王亚鑫,谢晖* ,黄莉,杨远正( 南京工业大学化工学院,南京210009)
UV 固化技术由于其低能耗、环境污染少、工艺成本低及室温快速固化等优点,近年来引起了越来越多研究者的关注。环氧丙烯酸酯是UV 固化涂料众多预聚物中应用最广泛的一种,其固化膜具有硬度高、附着力好、固化速度快及耐化学性好等优点; 但环氧丙烯酸树脂固化膜却存在韧性差、黏度高、脆性大及耐老化性差等缺点,大大限制了其应用。因此,为了改善环氧丙烯酸酯的性能,拓展它的应用范围,对其进行改性尤为重要。
目前,改性方法主要有物理改性和化学改性。物理改性是在紫外光固化有机体系中引入无机相,从而制备出有机-无机杂化材料,借助有机相和无机相的协同作用提升固化膜的某些性能。Liu 等通过光化学反应诱导溶胶-凝胶法制备紫外光固化环氧丙烯酸酯-二氧化硅杂化涂层,结果表明: 当复合涂层含有10%二氧化硅时,固化膜热稳定性最好。物理改性虽然简单有效,但杂化膜内容易发生相分离,难以得到均质膜。化学改性的方法之一是将环氧丙烯酸酯中的环氧基或开环后的羟基作为活性位点,与其他功能单体反应,向体系中引入软链或硬链结构,从而适当调节固化膜的柔韧性或硬度。Yan 等利用生物基材料木质素来改性环氧丙烯酸酯,研究发现木质素加入量为环氧树脂质量的10%时,木质素基环氧丙烯酸酯的涂膜硬度3H,附着力为1 级,柔韧性5 mm,然而在木质素分子结构中存在大量苯丙烷单元,柔韧性提升并不明显。王晓丽等将环氧树脂先与二元酸( 丁二酸、癸二酸、己二酸及辛二酸) 部分开环,再与丙烯酸酯化,制得柔韧性优异的环氧丙烯酸酯,虽然二元酸提升了涂膜韧性,但改性树脂黏度较大。韦星航等用含柔性链段的聚乙二醇二缩水甘油醚对环氧树脂进行降黏增韧改性,再用衣康酸进行封端,所得涂膜柔韧性、附着力及耐冲击性均有提升,但衣康酸分子结构含有碳碳双键结构,在较高温度下体系容易发生副反应,甚至凝胶。上述方法所合成的改性环氧丙烯酸酯,柔韧性得到不同程度的提高,但在制备过程存在易凝胶,反应时间长等问题; 改性树脂黏度也较高,需要加入较多活性稀释剂或溶剂; 此外,所合成改性环氧丙烯酸酯官能度较低。因此,为了充分满足改性环氧丙烯酸树脂生产安全要求、环保要求及UV 涂料市场需求,仍需对环氧丙烯酸酯进行复合改性研究。
本研究使用月桂二酸( DDDA) 与环氧树脂( E-51) 和1,4-丁二醇二缩水甘油醚( BDDGE) 反应,对环氧树脂进行改性,再用甲基丙烯酸酐( MA2O) 进行封端,制得低黏度柔性环氧丙烯酸树脂。一方面利用低黏度活性稀释剂BDDGE 代替部分环氧树脂,降低了体系黏度,同时将含有长链柔性链段的DDDA 和BDDGE 引入了基体树脂,通过内部增塑作用有效提高了环氧丙烯酸酯的柔性; 另一方面,利用反应活性较高的MA2O 向体系中引入碳碳双键,所得树脂结构不仅为端双键,还含有侧链的双键,具有更高的官能度。
1 实验
1. 1 主要原料及仪器
环氧树脂( E-51) : 工业级,南通星辰合成材料有限公司; 1,4-丁二醇二缩水甘油醚( BDDGE) : 工业级,上海迈瑞尔化学技术有限公司; 月桂二酸( DDDA) : 工业级,淄博广通化工责任有限公司; 甲基丙烯酸酐( MA2O) 、1,4-丁二醇二缩水甘油醚二丙烯酸酯: 实验室自制[10-11]; 四丁基溴化铵( TBAB) 、三苯基膦( TPP) 、N,N-二甲基苄胺( BDMA) : 分析纯,阿拉丁试剂有限公司; 对苯二酚( HQ) 、对羟基苯甲醚( MQ) : 分析纯,国药集团化学试剂有限公司; 三丙二醇二丙烯酸酯( TPGDA) : 工业级,长兴化学材料( 珠海) 有限公司; 光引发剂1173: 工业级,光易化工。
Nicolet 6700 型傅里叶红外光谱仪: 美国ThermoScientific 公司; WRT-2P 型热分析仪( TGA) : 上海精密科学仪器公司; UV 固化机: 深圳市能佳自动化设备有限公司; NDJ - 1 型旋转黏度仪: 上海天平仪器厂; QHQ 型涂膜铅笔划痕硬度仪、QFZ 型涂膜附着力试验仪、QTX 型漆膜柔韧性测定仪、QCJ 型漆膜冲击器: 天津精科材料试验机厂。
1. 2 改性环氧丙烯酸酯的合成
在配备有电动搅拌器、球形冷凝管及感温探头的四口烧瓶中,加入一定量的E-51、BDDGE、DDDA 及催化剂四丁基溴化铵,搅拌升温至90 ℃,开始保温。反应过程中,以0. 5 h 为间隔检测体系酸值。当体系的酸值小于5 mgKOH/g 时,将温度降至80 ℃,并向烧瓶中加入适量催化剂四丁基溴化铵和阻聚剂对羟基苯甲醚; 同时以每秒2~3 滴的速率滴加MA2O。将反应温度保持在80 ℃,直到体系酸值小于5 mgKOH/g时,停止反应并将温度迅速降至室温,即可得改性环氧丙烯酸酯( MEA) 。MEA 合成路线如式( 1) 所示。

1. 3 改性环氧丙烯酸酯涂膜的制备
将70%( 质量分数,下同) 合成的改性环氧丙烯酸酯、25%三丙二醇二丙烯酸酯( TPGDA) 、4%光引发剂1173 及1%助剂混合均匀后,用喷枪喷涂在已经处理过的马口铁片上,然后在70 ℃ 的烘箱中干燥5 min,最后置于UV 固化机中用紫外灯辐射固化成膜。
1. 4 分析测试
按GB /T 2895—2008 测定反应体系酸值; 按GB /T 9751. 1—2008 测定树脂黏度; 按GB /T 6753. 3—1986 测定树脂贮存稳定性; 按GB /T 1731—1993 测定漆膜的柔韧性; 按GB /T 6739—2006 测定漆膜的硬度; 按GB /T 9286—1998 测定漆膜的附着力; 按GB /T1732—1993 测试漆膜的耐冲击性; 按GB /T 1733—1993 测试漆膜耐水性。
采用傅里叶红外光谱仪对产物的结构进行表征,用KBr 压片,测定树脂的透光率,扫描范围4 000~400 cm-1。采用热重分析仪对涂膜的热稳定性进行测试。在N2气氛下,温度范围为30 ~ 600 ℃,升温速率为10 ℃ /min。
2 结果与讨论
2. 1 反应温度、反应时间对MEA 合成的影响
改性环氧丙烯酸酯的合成主要由以下两步组成;第一步,月桂二酸与环氧基团的开环反应; 第二步,甲基丙烯酸酐优先与体系中的羟基反应,生成的产物甲基丙烯酸再与环氧树脂反应,从而引入可光固化的双键结构,所得树脂不仅是端双键结构,还含有侧链的双键,具有更高的官能度。两步反应均属于酯化反应,实验中提高反应温度,能提高反应速率,缩短到达反应平衡的时间; 但温度太高时会引发体系的环氧基与侧链羟基的反应以及双键聚合等副反应的发生,并且过高的温度会导致反应过于剧烈而不易控制,从而发生凝胶。改性环氧丙烯酸树脂合成过程中副反应如式( 2) 、式( 3) 所示。因此,合适的反应温度对于反应安全高效的进行至关重要。

2. 1. 1 第一步反应温度和反应时间对产物性能的影响
以E-51、BDDGE 及DDDA 为原料,使用四丁基溴化铵作为催化剂,探讨反应温度和反应时间对第一步反应的影响,反应温度选取了85 ℃、90 ℃、95 ℃三个水平。表1 和图1 显示了第一步反应温度和时间对产物性能的影响。

从图1 可以发现,随着温度升高,反应速率加快,酸值达到相同值所需时间减少,主要原因是温度越高,分子热运动越快,碰撞几率增大,使反应速率加快; 通过比较90 ℃和95 ℃时温度和时间对酸值的影响,可以看出当反应达到一定温度时,再升高温度,对酸值影响不大,两者分别在2. 5 h 和2 h 反应达到终点; 由表1 发现,95 ℃时体系颜色加深,变为橙黄色,同时产物黏度很大,说明温度过高导致副反应增多,而且过高的温度易发生凝胶。所以,为了保证较高的酯化程度和较好的产品色泽,第一步最佳的反应条件为: 温度90 ℃,时间2. 5 h。
2. 1. 2 第二步反应温度和反应时间对产物性能的影响
在第二步反应中,MA2O 先与体系中的羟基反应,生成的产物甲基丙烯酸再与环氧基进行封端反应,同时引入光敏基团。实验中固定其他条件不变,选取70 ℃、80 ℃、90 ℃三个水平,考察反应温度和时间对反应的影响。表2 和图2 显示了第二步反应温度和时间对产物的影响。

从图2 可以看出,相对第一步反应,第二步反应对于反应温度的要求有所降低,在低于90 ℃的温度条件下,反应可在3 h 内达到平衡,其原因可能为MA2O,由于其独特的酸酐结构,使得在与环氧基反应时反应活性较高,对温度的要求较低,同时反应速率较快。由图2 可以发现在90 ℃条件下,酸值在较短时间达到较低水平,但相对于70 ℃和80 ℃,其体系黏度相对较高,而且在70 ℃下,随着反应时间的延长,酸值很难降至5 mgKOH/g 以下,不能满足工业应用要求。因此,为了保证较低的酸值和黏度,第二步最佳的反应条件为: 温度80 ℃,时间2. 5 h。
2. 2 催化剂对合成MEA 的影响
对于环氧丙烯酸酯的合成反应,常用的催化剂有季铵盐、叔胺盐及三苯基膦等。四丁基溴化铵是季铵盐中的一种,并且也是制备环氧丙烯酸酯常用的催化剂。下文以四丁基溴化铵为例,讨论其催化机理。四丁基溴化铵在加热条件下容易分解成叔胺和卤代烃;由于叔胺上的氮原子上有一对孤对电子,它易于对环氧基团进行亲核进攻,使之开环生成四丁基铵内盐;四丁基铵内盐随后吸收羧酸的质子形成铵盐和羧酸负离子; 最后与铵盐氮原子相近的碳原子具有亲电性,可与羧酸根负离子反应生成环氧丙烯酸酯,同时催化剂再生。
2. 2. 1 催化剂种类对酯化率的影响
固定第一步反应温度90 ℃,反应2. 5 h,第二步反应温度80 ℃,反应时间2. 5 h,催化剂用量0. 5%( 以反应前原料总质量计) 条件不变,考察催化剂三苯基膦( TPP) 、N,N-二甲基苄胺( BDMA) 及四丁基溴化铵( TBAB) 对第一步反应和第二步反应酯化率的影响,所得结果如图3 所示。酯化率的计算方法如式( 4) 所示。

式中: X0—反应初始的酸值; Xt—即时检测的酸值。

由图3( a) 可知,催化剂的种类对第一步反应速率影响不大,使用不同催化剂进行实验,最终反应酯化率相差不大,N,N-二甲基苄胺的催化性能相比四丁基溴化铵来说更好一些,同时,三苯基膦相对上述2 种催化剂的催化效果则较差。然而,催化剂种类对第二步反应却有着较大影响。由图3( b) 可以发现,相对于另外2 种催化剂反应体系使用四丁基溴化铵作为催化剂,催化性能得到明显提高,N,N-二甲基苄胺及三苯基膦在第二步反应中,具有较低的催化活性。考虑到第二步反应对产品的合成更为重要,因为其决定着最终的光固化性能及产品是否达标。因此,反应过程中,使用四丁基溴化铵作为反应催化剂较为合适。
2. 2. 2 催化剂用量对酯化率的影响
对于聚合反应来说,合适的催化剂用量可以有效降低反应活化能,增大反应速率。催化剂用量太少,会造成聚合反应时间较长,并且发生醚化等副反应的几率增加; 过量的催化剂则可能引起局部反应温度过高,导致聚合反应不均一、副反应增加。为了考察催化剂用量对酯化率的影响,以四丁基溴化铵( TBAB)作为催化剂,控制其他条件不变,探索了催化剂用量( 以反应前原料总质量计) 为0. 1%、0. 2%、0. 3%、0. 4%、0. 5%、0. 6%时的酯化率。结果如图4 所示。

由图4 可以看出,在第一步反应中,加入少量催化剂即可快速达到平衡,在催化剂加入量为0. 3%时,继续增加催化剂用量,酯化率很难继续提升,原因可能为在月桂二酸与环氧树脂的开环过程中,月桂二酸与环氧树脂均为二官能单体,同时为了下一步的引入双键,环氧树脂只需部分开环,理论上能够生成大分子链,所以加入少量催化剂即可达到反应平衡。在第二步反应中,则需相对较多的催化剂才能实现较高的酯化率; 当催化剂加入量为0. 5%时,反应达到平衡,当催化剂为0. 6%时,体系颜色开始变深。
综上所述,第一步反应选择加入0. 3%的催化剂,第二步反应选择加入0. 5%的催化剂。
2. 3 阻聚剂对合成MEA 的影响
在实际生产中,选择合适的阻聚剂对树脂的稳定、贮存及运输都有很大的好处。在上述研究的基础上,控制其他条件不变,考察酚类阻聚剂对苯二酚和对羟基苯甲醚对MEA 树脂合成反应的影响,实验所得结果如表3 所示。

由表3 可以看出,使用对苯二酚作为阻聚剂,随着对苯二酚加入量的增加,产品的颜色由淡黄透明逐渐变为橙黄半透明,这是因为对苯二酚含有苯环结构,易黄变。另外,对苯二酚在加入量较多时才能发生较好的阻聚效果。然而,对羟基苯甲醚与对苯二酚不同,随着对羟基苯甲醚加入量的加大,对产品的色泽并不没有发生太大的影响,而且在较低的加入量时依然保持了较好的阻聚性能。同时,考虑到阻聚剂加入过多,在后续光固化成膜时,容易造成固化速度慢,固化不彻底的情况。综合比较,使用对羟基苯甲醚作为阻聚剂,加入量为0. 06%( 以反应前原料总质量计) 最为合适。
2. 4 红外表征
图5 为E-51 和MEA 的红外光谱。
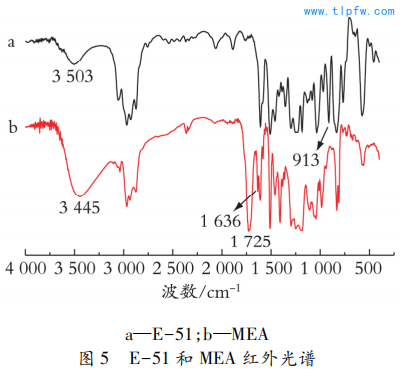
由图5 可知,MEA 在3 445 cm-1处的羟基吸收峰明显加强,913 cm-1 处环氧特征峰消失,说明环氧树脂的环氧基团发生开环反应,打开了环氧键,使得MEA 比E-51 有更多的羟基; 2 968 cm-1、2 876 cm-1为甲基和亚甲基的C—H伸缩振动吸收峰; 1 725 cm-1处出现强烈的吸收峰,为酯羰基C ?O的吸收峰,说明产物存在酯基结构; 1 636 cm-1出现C ?C伸缩振动峰,说明碳碳双键已成功引入至预聚体主链上; 酸酐在1 786 cm-1处的羰基C ?O伸缩振动吸收峰消失; 同时,E-51 的某些特征吸收峰在MEA 谱图中得到保留,其中1 608 cm-1为芳环C ?C伸缩振动吸收峰,1510 cm-1、831 cm-1 为对位取代的苯环吸收峰,1 250 cm-1为脂肪芳香醚键吸收峰。因此,通过红外光谱对比分析可知成功合成出了目标产物MEA树脂。
2. 5 BDDGE 用量对树脂黏度及涂膜性能的影响
本实验采用BDDGE 对E-51 进行改性,研究了BDDGE 与E-51 的物质的量比对合成树脂黏度及涂膜性能影响,结果如表4 所示。

BDDGE 是一种长链双官能度环氧活性稀释剂,也是环氧树脂的增韧稀释剂。由表4 可以看出,随着BDDGE 添加量的增加,MEA 的黏度显著下降; 与未加BDDGE 改性时相比,当BDDGE 与E-51 物质的量比为0. 6 时,所合成的MEA 树脂黏度为15 060 mPa·s,降黏率达到37%,原因可能为BDDGE 分子主要由柔性链—C—O—C—构成,本体黏度低,将其引入到E-51 中,使体系旋转活化能降低,分子链流动性增强,所以降黏显著。同时,随着BDDGE 添加量的增加,涂膜铅笔硬度变化较小,但附着力、柔韧性及耐冲击性均有明显提升,说明引入BDDGE 提高了涂膜的综合性能; 这是因为BDDGE 含有2 个环氧基团,固化时均参与反应,可形成链状及网状结构,固化后可大大提高与基材的附着力、柔韧性及耐冲击性,改善涂膜的脆裂开裂等缺点。当BDDGE 和E-51 的物质的量比为0. 6 时,涂膜综合性能优良。
2. 6 改性环氧丙烯酸酯与混合环氧丙烯酸酯涂膜性能对比
本实验比较了改性环氧丙烯酸酯与混合环氧丙烯酸酯( 1,4-丁二醇二缩水甘油醚二丙烯酸酯和双酚A 环氧丙烯酸酯物质的量比为0. 6 时) 固化涂膜的性能,结果如表5 所示。

由表5 可知,改性环氧丙烯酸酯与混合环氧丙烯酸酯固化膜性能相比,附着力、柔韧性及耐冲击性均有提高,但硬度并无变化。这是由于长碳链月桂二酸不含碳碳双键,使体系的双键含量减少,体系固化收缩减少,附着力得到提高; 同时,体系中柔性链段月桂二酸和1,4-丁二醇二缩水甘油醚的引入,增加了内旋转的单键数量,固化膜的柔韧性与耐冲击性得到改善,但正由于两者的引入,使得改性环氧丙烯酸树脂分子链结构中所含刚性基团苯环的比例降低,从而导致固化膜的硬度没有得到提升。改性环氧丙烯酸酯耐水性略胜一筹,原因可能为疏水性脂肪族长链1,4-丁二醇二缩水甘油醚的引入,使得树脂与水的氢键作用、电子效应减弱,耐水性能得到提高。
2. 7 热重分析
图6 为改性环氧丙烯酸酯( a) 与混合环氧丙烯酸酯( b) 固化膜的热重曲线。

由图6 可知,随着温度的升高,( a) 和( b) 固化膜质量均逐渐减少,并且热失质量曲线的变化趋势基本相同; 但对比发现,当失去相同质量分数时,( b) 的分解温度总比( a) 高。( a) 的总热失质量率为86. 05%,高于( b) 的83. 44%; 同时,( a) 和( b) 热分解温度( 失质量5%的温度) 分别为243 ℃、253 ℃,表明自制改性环氧丙烯酸酯的热稳定性有所下降,这可能是因为体系中含有了较多的柔性链段,使得刚性基团苯环的比例下降,进而使体系交联密度降低,最终影响了改性环氧丙烯酸酯的耐热性。
3 结语
( 1) 利用E-51、BDDGE、DDDA 及MA2O 等原料合成出了低黏度柔性环氧丙烯酸酯,合成工艺简单。两步反应的适宜条件为: 第一步反应温度90 ℃,反应时间2. 5 h,催化剂四丁基溴化铵用量为0. 3%; 第二步反应温度80 ℃,反应时间2. 5 h,催化剂四丁基溴化铵用量为0. 5%,阻聚剂对羟基苯甲醚用量为0. 06%。结果表明MA2O 作为封端剂,由于其独特的酸酐结构,可在较低温度发生酯化反应,体系发生凝胶的可能性大大降低,同时所得树脂具有更高的官能度。
( 2) BDDGE 对合成树脂的降黏效果较好,与未加BDDGE 相比,当BDDGE 和E-51 的物质的量比为0. 6 时,树脂降黏率达到37%,同时涂膜性能优良。
( 3) 利用DDDA 中的羧基与环氧基发生开环反应,向E-51 中引入DDDA、BDDGE 结构中脂肪长链柔性链段,有效提高了环氧丙烯酸酯的柔韧性; 涂膜铅笔硬度为3H,附着力为1 级,柔韧性1 mm,耐冲击性50 cm,涂层耐水性较好,耐热性有所下降。





